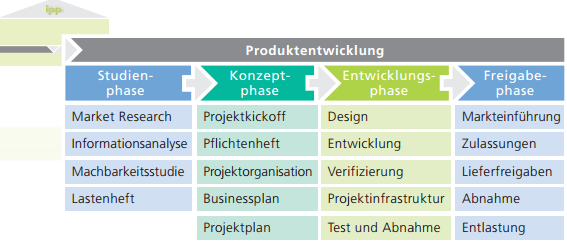
Marcelo Lackner
MDR und IEC 60601-1
Manager
Medical Device Expert & Regulations

Marcelo Lackner, Spezialist für MDR und IEC 60601-1, ist Diplom Ingenieur (FH) für Feinwerk- und Mikrotechnik. Den Schwerpunk legte er dabei auf die Medizintechnik.
Sein umfassendes und tiefes Wissen rund um Medizinprodukte, erwarb Herr Lackner bei den Benannten Stellen. Hierbei prüfte er im Labor verschiedene Geräte nach IEC 60601-1. Zusätzlich ist Marcelo Lackner ein ausgebildeter Auditor. Dies bietet einen zusätzlichen Nutzen für seinen Einsatz als Experte für MDR und IEC 60601-1.
Des Weiteren ist Herr Lackner als Experte für die Zulassung von Medizinprodukten im nationalen und internationalen Mark tätig. Daneben besitzt Herr Lackner mehr als 5 Jahre klinische Erfahrung und 10 Jahre Erfahrung in der Zulassung von Medizinprodukten.
Herr Lackner ist ein Technical Communicator Professional Level. Dafür absolvierte er im Jahr 2022 eine Ausbildung zum Technischen Redakteur bei der Gesellschaft für Technische Kommunikation – tekom Deutschland e.V.
Aktuell unterstützt Herr Lackner unsere Kunden bei der elektrischen Sicherheitsprüfung nach IEC 60601-1 und der entsprechenden partikulären Norm IEC 80601-2-60. Außerdem umfasst seine Tätigkeit das Sichten und Bewerten von Technischen Dokumentationen. Insbesondere für die Zulassung von Klasse Ir Medizinprodukten nach MDR und die Zulassung von chirurgischen Instrumenten für die Ophthalmologie. Zusätzlich beschäftigt sich Herr Lackner mit der Normenrecherche zur Vervollständigung von den GSPR. Außerdem steht er immer seinen Kollegen hilfsbereit zur Seite. Dabei übernimmt er zum Beispiel die Reviews von TechDok Dokumenten.
Marcelo Lackner ist Ihr Spezialist für die technische Dokumentation nach MDR, Regulatory Affairs sowie IEC 60601-1 am Standort Nürnberg in Bayern.
 Regulatory Affairs Medizintechnik
Regulatory Affairs Medizintechnik