Technische Dokumentation: Umstellung MDD auf MDR (Low Budget)
IPP Notizen aus der Praxis:
Ausgangslage des Kunden
Ein Kleinstunternehmen benötigt fachliche und personelle Unterstützung bei der Anpassung der Technischen Dokumentation an die Anforderungen MDR (Medical Device Regulation, 2017/745).
Es handelt sich um bisher nach MDD (93/42/EWG) zugelassene Medizinproduktegruppe der Klasse IIa.
Die Umstellung MDD auf MDR soll mit minimalistischem Aufwand (Low Budget) erfolgen.
Das Projekt soll innerhalb von drei Monaten abgeschlossen sein.
Durchführung der Umstellung MDD auf MDR:
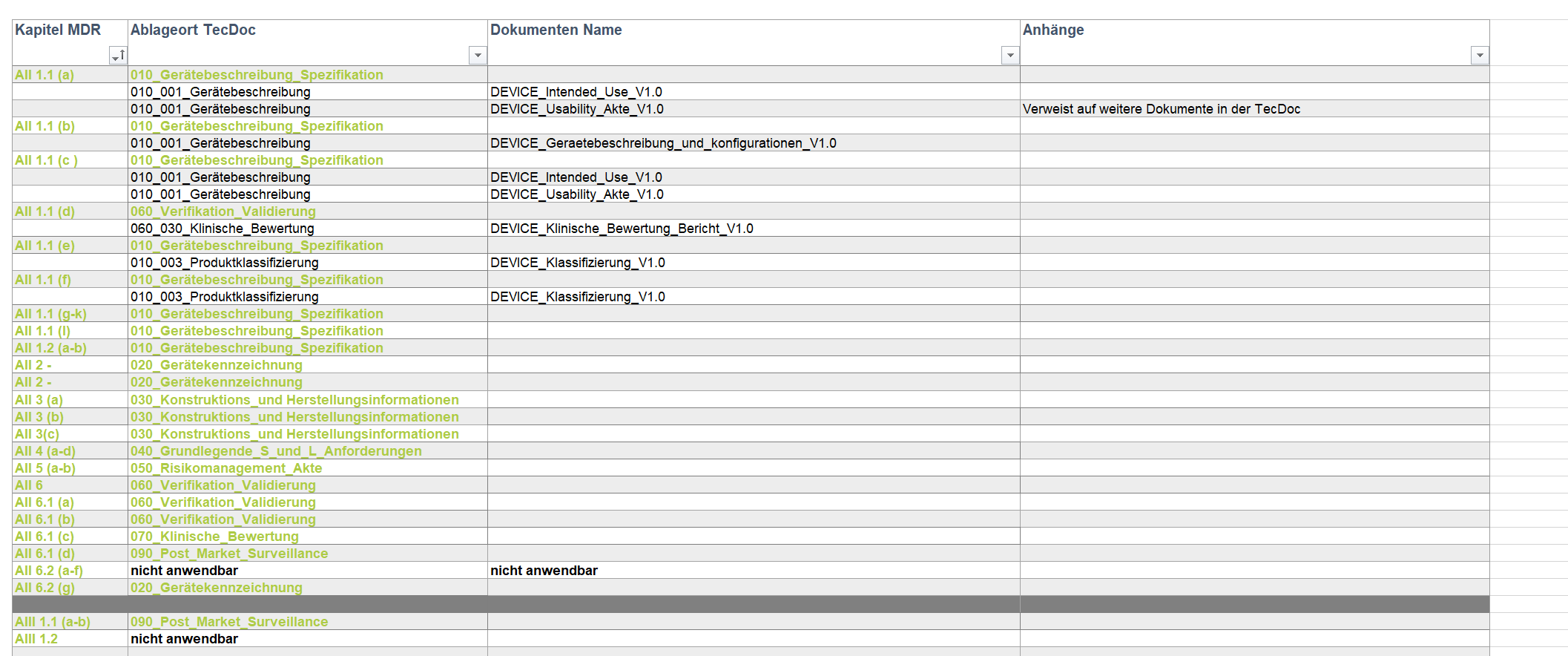
- Gap-Analyse der bestehenden Technischen Dokumentation (TecDoc) nach MDD (93/42/EWG) zugelassenen Klasse IIa Medizinproduktegruppe, um den Aufwand der Unterstützung bei der Umsetzung auf MDR zu berechnen.
- Umstrukturierung der bestehenden TecDoc an die Strukturvorgabe der benannten Stelle.
- Neuerstellungen und Anpassung der TecDoc gemäß Gap-Analyse.
- Übergabebesprechung mit Vorstellung der neuen TecDoc Struktur und der Dokumente sowie Auflistung der finalen Aufgaben des Herstellers.
Ergebnis
Wir konnten das Projekt termingerecht abschließen.
Der Kunde kann mit unserer Unterstützung die geforderte Technische Dokumentation nach MDR der benannten Stelle termingerecht übergeben und kann die TecDoc selbstständig weiter pflegen.
Fragen?
Sie benötigen Unterstützung oder Beratung zum Thema Technische Dokumentation von Medizinprodukten sowie der Umstellung MDD auf MDR durch unsere Experten? Sie erreichen uns telefonisch unter : +49 911 36069710 sowie per Email unter kontakt(at)ipp-nbg.com
Bei Fragen rund um Ihr Medizinprodukt:
Unser erfahrenes sowie vielseitiges Team berät Sie gerne. Wir navigieren und unterstützen Sie auf dem Weg durch den Dschungel der Regularien. Die Anforderungen an Hersteller von Medizinprodukten steigen immer weiter und wir behalten für Sie den Überblick. Ihre Vorteile:
- Sie haben Termine und Kosten im Griff
- Wir sind up to date bei den Regularien: MDR sowie der geltenden Medizingesetze und -Richtlinien
- Sie nutzen ein qualifiziertes sowie erfahrenes Team
- Sie erhalten alles aus einer Hand
- Des Weiteren entscheiden Sie selbst, wieviel Sie sich einbringen möchten