Maren Fiedler
Maren Fiedler ist Consultant bei ipp. mit Schwerpunkt Technische Dokumentation nach MDR. Sie verbindet technisches Verständnis mit regulatorischem Projektmanagement und begleitet Projekte in Nürnberg.

Maren Fiedler ist Consultant bei ipp. mit Schwerpunkt Technische Dokumentation nach MDR. Sie verbindet technisches Verständnis mit regulatorischem Projektmanagement und begleitet Projekte in Nürnberg.
Corinna Locke ist Consultant bei ipp. im Bereich Regulatory Projektmanagement. Sie unterstützt Kunden bei Strategie, Compliance und Technischer Dokumentation für Medizinprodukte.

Georgiana Niedermaier unterstützt bei ipp. Projekte rund um Projektmanagement, Prozessberatung und Technische Dokumentation. Ihr Fokus liegt auf ISO 13485/MDR und sauberer, umsetzbarer Dokumentation.

Nadine Pauli unterstützt bei ipp. die Technische Dokumentation nach MDD/MDR. Zusätzlich arbeitet sie im Regulatory Projektmanagement für planbare, compliance-sichere Ergebnisse.

Alexander Nellner ist Manager für regulatorische Anforderungen und Technische Dokumentation. Sein Schwerpunkt liegt auf klarer Nachweisführung, belastbaren Produktakten und sauber strukturierten MDR-Projekten.

Gabriela Rödig stärkt bei ipp. die technische Kommunikation. Sie sorgt dafür, dass komplexe Inhalte klar, konsistent und für Projekte sowie Dokumentationen gut nutzbar sind.

Andre Çakici unterstützt bei ipp. die Technische Dokumentation nach MDR und das Regulatory Projektmanagement. Sein Fokus liegt auf sauberer Struktur, Nachweisen und effizienter Umsetzung.

Dr. Volker Klügl verantwortet anspruchsvolle MedTech-Projekte an der Schnittstelle von Strategie, Regulatory und Umsetzung. Er steht für belastbare Nachweisführung, klare Projektlogik und einen Arbeitsstil, der moderne Werkzeuge und KI dort intelligent einsetzt, wo sie Struktur, Tempo und Entscheidungsqualität sinnvoll verbessern.

Welche UDI-Frist gilt wann? Diese Übersicht zeigt die MDR-Kennzeichnungsfristen je nach Produktklasse und je nachdem, ob der UDI-Träger auf Verpackung oder direkt am Produkt angebracht wird.

Ein Kleinstunternehmen benötigt fachliche und personelle Unterstützung bei der Anpassung der Technische Dokumentation an die Anforderungen MDR (Medical Device Regulation, 2017/745).
Praxisbericht zur Umstellung von MDD auf MDR: Wir zeigen, wie klinische Bewertungen für bereits zugelassene Produkte strukturiert und durchgeführt werden – von CEP und CER über State of the Art bis zur finalen Bewertung nach MDR (EU 2017/745) und MEDDEV 2.7/1 Rev.4.
Dr. Hofmann – Klinische Bewertungen MEDDEV 2.7/1 Revision 4 / MDR | Regulatory Projekt Management

M. Schön – Großprojekte | Technische Dokumentation nach 93/42/EWG / MDR | Prozessberatung ISO 13485 / MDR

Bettina Nachmann unterstützt bei ipp. als QMB und im Regulatory Service. Sie führt interne Audits durch und arbeitet an klinischen Bewertungen nach MEDDEV/MDR für ein stabiles ISO-13485-System.

Felix Nickel verantwortet bei IPP als PO die Redaktion von Easy13485. Er steht für strukturierte Umsetzung, technische Klarheit und belastbare Projektsteuerung in anspruchsvollen MedTech-Vorhaben.

Was ist aber, wenn Ihr OEM z.B. in China sitzt und “Ihr(e)” Medizinprodukt(e) oder Komponente nur eine chinesische TecDoc besitzt? Müssen sämtliche Dokumente übersetzt werden?
In den Anhängen II und III der MDR wird erläutert, was die Technische Dokumentation mindestens beinhalten soll. In Anhang II wird hierbei auf das Produkt selbst eingegangen und Anhang III legt die Anforderungen an die Marktüberwachung nach dem Inverkehrbringen mit Planung und Durchführung fest.
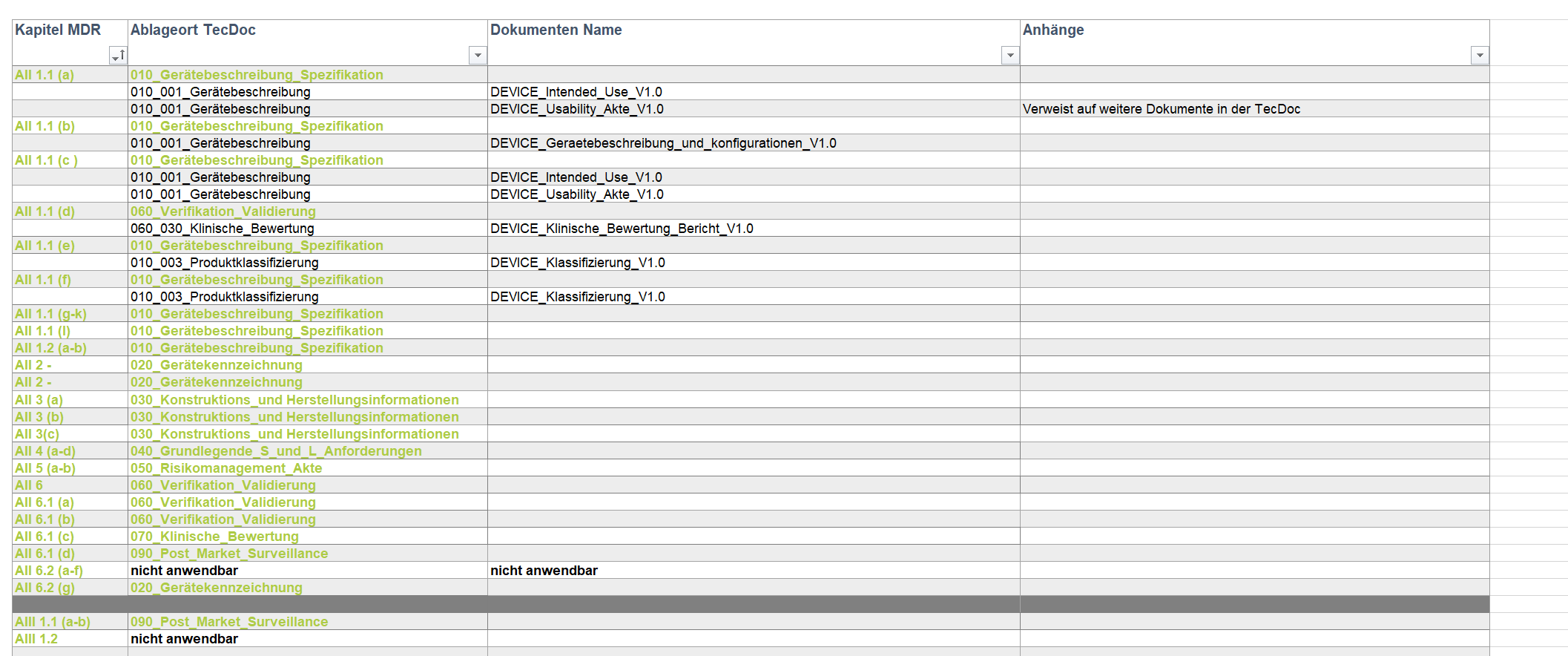
ipp. Notizen aus der Praxis: Bestehende Technische Dokumentation eines nach MDD zugelassen Medizinproduktes auf die MDR umstellen
Betrachtung verschiedener Konstellationen von Hersteller zu Lieferant nach MDR und 93/42/EWG, wie PLM OEM, Handelsware, Aufbringung von Markenlogos, Exklusivität
