Moritz Nickel
Moritz Nickel unterstützt bei ipp. Projektmanagement und Technische Dokumentation. Ein Schwerpunkt ist Requirements Engineering nach IEC 62304 für Software in Medizinprodukten.

Moritz Nickel unterstützt bei ipp. Projektmanagement und Technische Dokumentation. Ein Schwerpunkt ist Requirements Engineering nach IEC 62304 für Software in Medizinprodukten.
Alexander Nellner ist bei ipp. Experte für Technische Dokumentation und regulatorische Anforderungen. Sein Fokus liegt auf strukturierter, belastbarer Nachweisführung in der Produktakte.

ipp. unterstützte die Erstellung der Technischen Dokumentation nach MDR für einen neuen XRay-Sensor. Innerhalb von neun Monaten wurde eine normkonforme Produktakte erstellt, die die CE-Konformität nach MDR 2017/745 für die Erprobung des Sensors ermöglichte.
Zulassung von Medizinprodukten in Saudi-Arabien IPP Notizen aus der Praxis: Für die Zulassung von Medizinprodukten in Saudi-Arabien ist die SFDA …

Welche UDI-Frist gilt wann? Diese Übersicht zeigt die MDR-Kennzeichnungsfristen je nach Produktklasse und je nachdem, ob der UDI-Träger auf Verpackung oder direkt am Produkt angebracht wird.

Ein Kleinstunternehmen benötigt fachliche und personelle Unterstützung bei der Anpassung der Technische Dokumentation an die Anforderungen MDR (Medical Device Regulation, 2017/745).
Praxisbericht zur Umstellung von MDD auf MDR: Wir zeigen, wie klinische Bewertungen für bereits zugelassene Produkte strukturiert und durchgeführt werden – von CEP und CER über State of the Art bis zur finalen Bewertung nach MDR (EU 2017/745) und MEDDEV 2.7/1 Rev.4.
Bettina Nachmann unterstützt bei ipp. als QMB und im Regulatory Service. Sie führt interne Audits durch und arbeitet an klinischen Bewertungen nach MEDDEV/MDR für ein stabiles ISO-13485-System.

Was ist aber, wenn Ihr OEM z.B. in China sitzt und “Ihr(e)” Medizinprodukt(e) oder Komponente nur eine chinesische TecDoc besitzt? Müssen sämtliche Dokumente übersetzt werden?
In den Anhängen II und III der MDR wird erläutert, was die Technische Dokumentation mindestens beinhalten soll. In Anhang II wird hierbei auf das Produkt selbst eingegangen und Anhang III legt die Anforderungen an die Marktüberwachung nach dem Inverkehrbringen mit Planung und Durchführung fest.
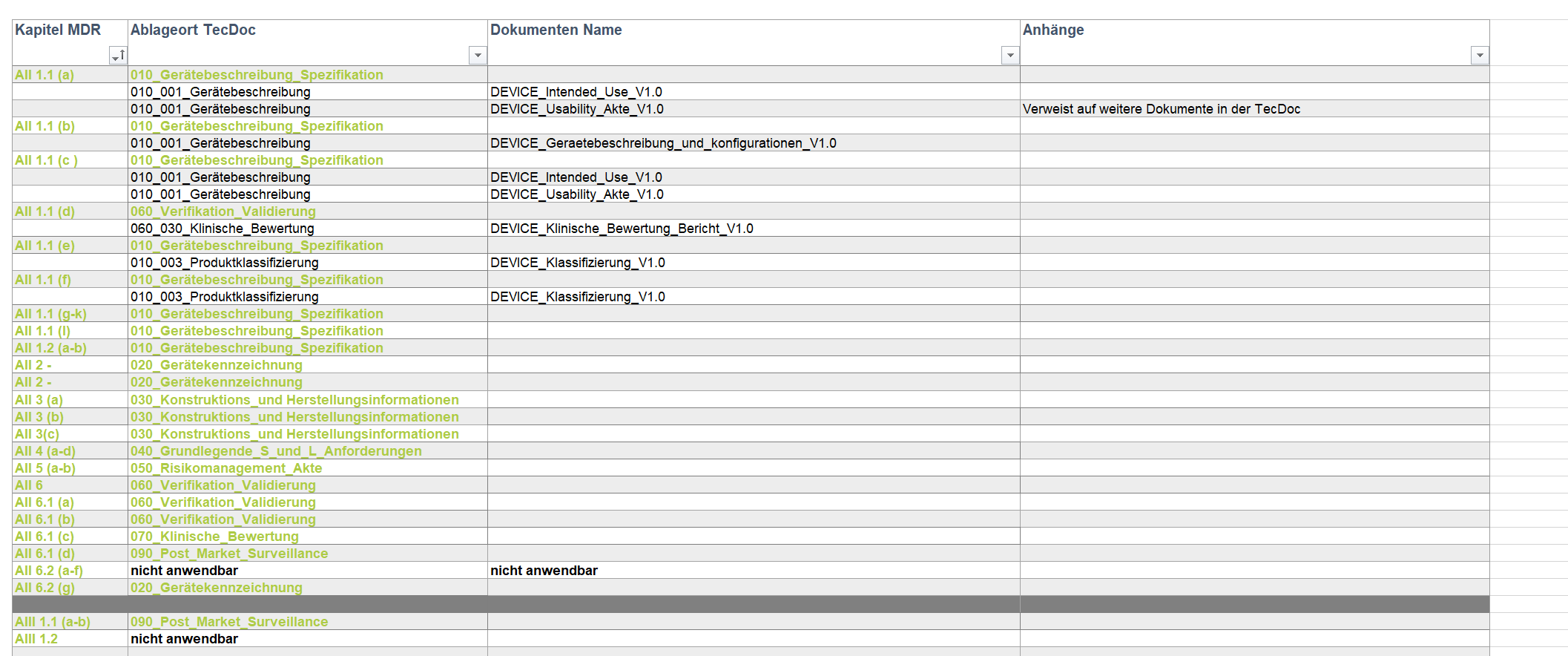
ipp. Notizen aus der Praxis: Bestehende Technische Dokumentation eines nach MDD zugelassen Medizinproduktes auf die MDR umstellen
ipp. Notizen aus der Praxis: Verifizierung und Validierung eines Medizinproduktes nach DIN EN 60529, DIN EN 13060 und ISO 15883 und technische Dokumentation nach MDD 93/42/EWG
ipp. Notizen aus der Praxis: Erfolgreiches Interimsmanagement für einen Hersteller von Medizinprodukten.
ipp. Notizen aus der Praxis: Erfolgreiches Einbinden von Testspezifikationen in ein SW-Tool zur Systemteststeuerung und -durchführung.
ipp. Notizen aus der Praxis: Import einer Produktakte in die ALM Software Polarion und Herstellen eines lückenlosen Tracings.
ipp. Notizen aus der Praxis: Programmieren einer Datenbank zur Erstellung eines neuen produktspezifischen Biokompatibilitätsnachweis.
ipp. Notizen aus der Praxis: Erfolgreiche Zulassung von Medizinprodukten auf internationalen Märkten.
