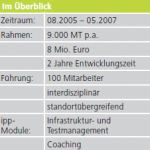
Medizinprodukt: PMO und Technische Dokumentation nach Richtlinie 93/42/EWG
ipp. Notizen aus der Praxis: Erfolgreiches Interimsmanagement für einen Hersteller von Medizinprodukten.
Unsere Leistungen Medizintechnik umfassen die strukturierte Unterstützung von Herstellern von Medizinprodukten entlang des gesamten Produktlebenszyklus.
ipp. Notizen aus der Praxis: Erfolgreiches Interimsmanagement für einen Hersteller von Medizinprodukten.
ipp. Notizen aus der Praxis: Erfolgreiche Unterstützung bei der Zertifizierung nach ISO 9001.
ipp. Notizen aus der Praxis: Erfolgreicher Aufbau und Optimierung eines Premium-Supports für ein komplexes Produkt aus der Dentalmedizin.
ipp. Notizen aus der Praxis: Durchführung eines internen Audits bei einem Hersteller für Medizinprodukte.

ipp. Notizen aus der Praxis: Erfolgreiches Einbinden von Testspezifikationen in ein SW-Tool zur Systemteststeuerung und -durchführung.
ipp. Notizen aus der Praxis: Import einer Produktakte in die ALM Software Polarion und Herstellen eines lückenlosen Tracings.
Praxisbericht: Zwei Produktfamilien wurden nach MEDDEV 2.7/1 Rev. 4 aktualisiert – mit klarer Vorbereitung, systematischer Literaturbewertung, Äquivalenzprüfung und termingerechter Fertigstellung für die Benannte Stelle.
ipp. Notizen aus der Praxis: Programmieren einer Datenbank zur Erstellung eines neuen produktspezifischen Biokompatibilitätsnachweis.
ipp. Notizen aus der Praxis: Erfolgreiche Zulassung von Medizinprodukten auf internationalen Märkten.
ipp. Notizen aus der Praxis: Polarion als webbasierte ALM Software wird bei einem Medizintechnik Unternehmen eingeführt.
ipp. Notizen aus der Praxis: Erfolgreicher Launch eines Medizinprodukts auf internationalem Markt.
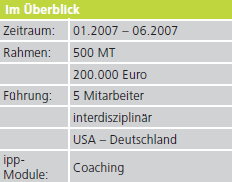
ipp. Notizen aus der Praxis: Erfolgreiches Interimsmanagement bei der Entwicklung eines Medizinprodukts.