In welcher Sprache muss eine Technische Dokumentation nach MDR vorliegen?
Was ist aber, wenn Ihr OEM z.B. in China sitzt und “Ihr(e)” Medizinprodukt(e) oder Komponente nur eine chinesische TecDoc besitzt? Müssen sämtliche Dokumente übersetzt werden?
Unsere Leistungen Medizintechnik umfassen die strukturierte Unterstützung von Herstellern von Medizinprodukten entlang des gesamten Produktlebenszyklus.
Was ist aber, wenn Ihr OEM z.B. in China sitzt und “Ihr(e)” Medizinprodukt(e) oder Komponente nur eine chinesische TecDoc besitzt? Müssen sämtliche Dokumente übersetzt werden?
In den Anhängen II und III der MDR wird erläutert, was die Technische Dokumentation mindestens beinhalten soll. In Anhang II wird hierbei auf das Produkt selbst eingegangen und Anhang III legt die Anforderungen an die Marktüberwachung nach dem Inverkehrbringen mit Planung und Durchführung fest.
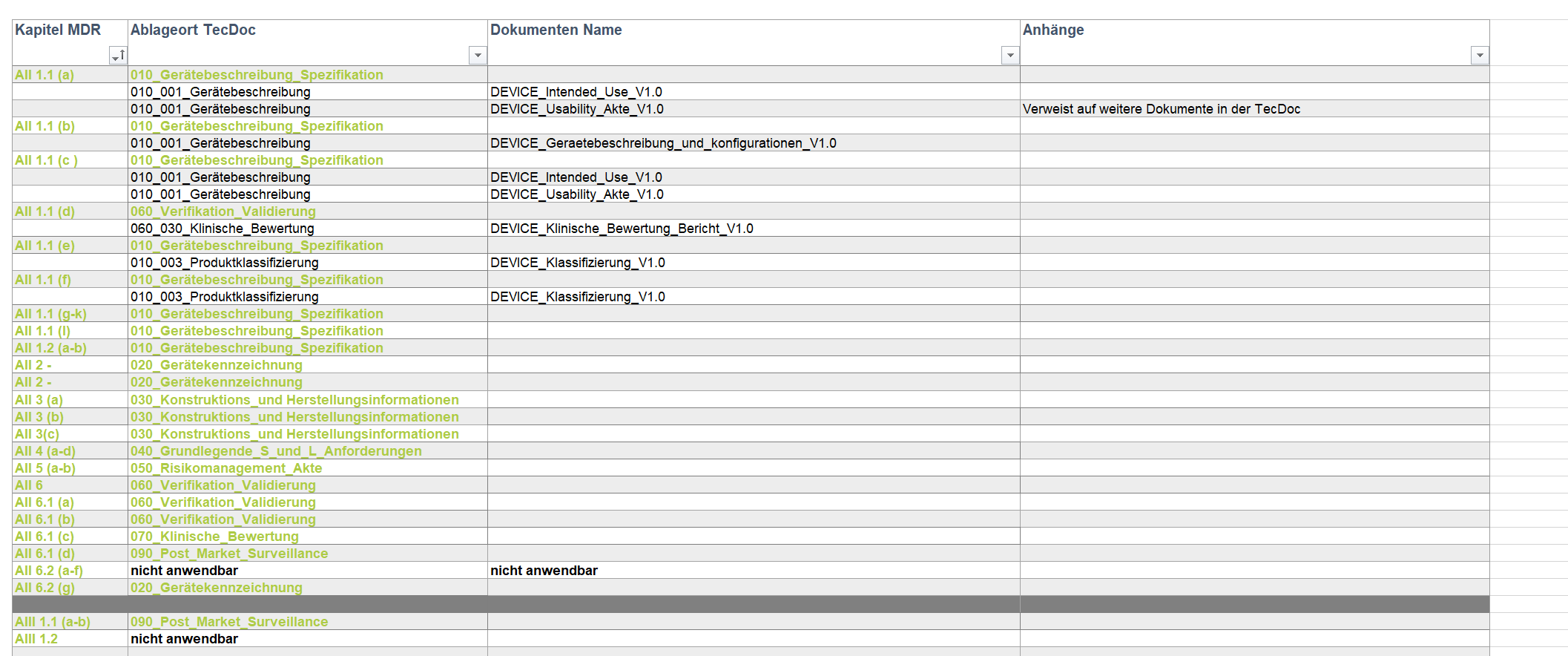
ipp. Notizen aus der Praxis: Bestehende Technische Dokumentation eines nach MDD zugelassen Medizinproduktes auf die MDR umstellen
Marktüberwachung braucht Daten aus der Anwendung – aber DSGVO-konform. So grenzen Sie Kundenbefragungen von klinischen Studien ab, vermeiden personenbezogene Daten (z. B. in Freitextfeldern) und bleiben gleichzeitig meldepflicht-sicher.
Betrachtung verschiedener Konstellationen von Hersteller zu Lieferant nach MDR und 93/42/EWG, wie PLM OEM, Handelsware, Aufbringung von Markenlogos, Exklusivität
ipp. Notizen aus der Praxis: Verifizierung und Validierung eines Medizinproduktes nach DIN EN 60529, DIN EN 13060 und ISO 15883 und technische Dokumentation nach MDD 93/42/EWG
ipp. Notizen aus der Praxis: Erfolgreiche Prozessoptimierung bei der Entwicklung von Medizinprodukten durch die Harmonisierung von Anforderungen aus verschiedenen Normen.
Martkzugang USA Freigabe, der US-Arzneimittelbehörde (U.S. Food and Drug Administration, FDA)
ipp. Notizen aus der Praxis: Beispiel für erfolgreiche Umsetzung eines externen QMB für einen Hersteller von Medizinprodukten für ISO 13485
Die Validierung von Prozesssoftware ist ein zentraler Bestandteil der normkonformen Herstellung von Medizinprodukten. Der Beitrag beschreibt regulatorische Anforderungen nach ISO 13485 und EU-MDR sowie typische Vorgehensweisen aus der Praxis.
Die BVMed Broschüre zu Konformitätsbewertungsverfahren für Medizinprodukte richtet sich an Hersteller und Verantwortliche. ipp. empfiehlt den Leitfaden als grundlegende Orientierung zur Einordnung regulatorischer Pflichten und Verantwortlichkeiten.
Die BVMed Klassifizierungsliste für Medizinprodukte dient Herstellern als Orientierungshilfe bei der Klassifizierung ihrer Produkte. ipp. empfiehlt den Leitfaden als Grundlage für die Wahl des passenden Konformitätsbewertungsverfahrens und die CE-Kennzeichnung.
Die Selbstklassifizierung von Medizinprodukten der Klasse 1 liegt in der Verantwortung des Herstellers. ipp. empfiehlt den vom BVMed verlinkten EU-Leitfadenentwurf als praxisnahe Orientierung zur MDR-konformen Klassifizierung.
Die BVMed Broschüre „Benannte Stellen“ richtet sich an Hersteller von Medizinprodukten mit externer Zertifizierungspflicht. ipp. empfiehlt den Leitfaden als fundierte Orientierung zu Aufgaben, Zuständigkeiten und zum Wechsel Benannter Stellen.
Unsere Leseempfehlung: Die BVMed-Broschüre zur klinischen Bewertung erklärt kompakt, warum die klinische Bewertung ein Kernbaustein für den Marktzugang ist – und worauf Hersteller in der Praxis achten sollten
Die BVMed Broschüre zum Risikomanagement für Medizinprodukte basiert auf praktischen Industrieerfahrungen. ipp. empfiehlt den Leitfaden als Orientierungshilfe für Hersteller im Umgang mit Normen, Benannten Stellen und Behörden.
Die BVMed Broschüre zur Marktüberwachung von Medizinprodukten richtet sich an alle Beteiligten im Umgang mit Vorkommnissen. ipp. empfiehlt den Leitfaden als praxisnahe Orientierung zur Umsetzung der Medizinprodukte-Sicherheitsplanverordnung.
Der BVMed-Leitfaden zum Off Label Use von Medizinprodukten unterstützt Hersteller beim Umgang mit Anwendungen außerhalb der Zweckbestimmung. ipp. empfiehlt die Broschüre als praxisnahe Orientierung zur Risikominimierung und regulatorischen Einordnung.
Der BVMed-Leitfaden zur Kennzeichnung von Medizinprodukten bietet Herstellern eine strukturierte Orientierung zu Kennzeichnungsanforderungen nach MDD und einschlägigen europäischen Normen. ipp. empfiehlt die Broschüre als grundlegende Informationsquelle.
Der BVMed-Leitfaden „Datenschutz bei Medizinprodukten“ unterstützt Hersteller und Akteure im Gesundheitswesen beim rechtssicheren Umgang mit personenbezogenen Daten nach EU-DSGVO und BDSG-neu. ipp. empfiehlt die Broschüre als praxisnahe Orientierung.