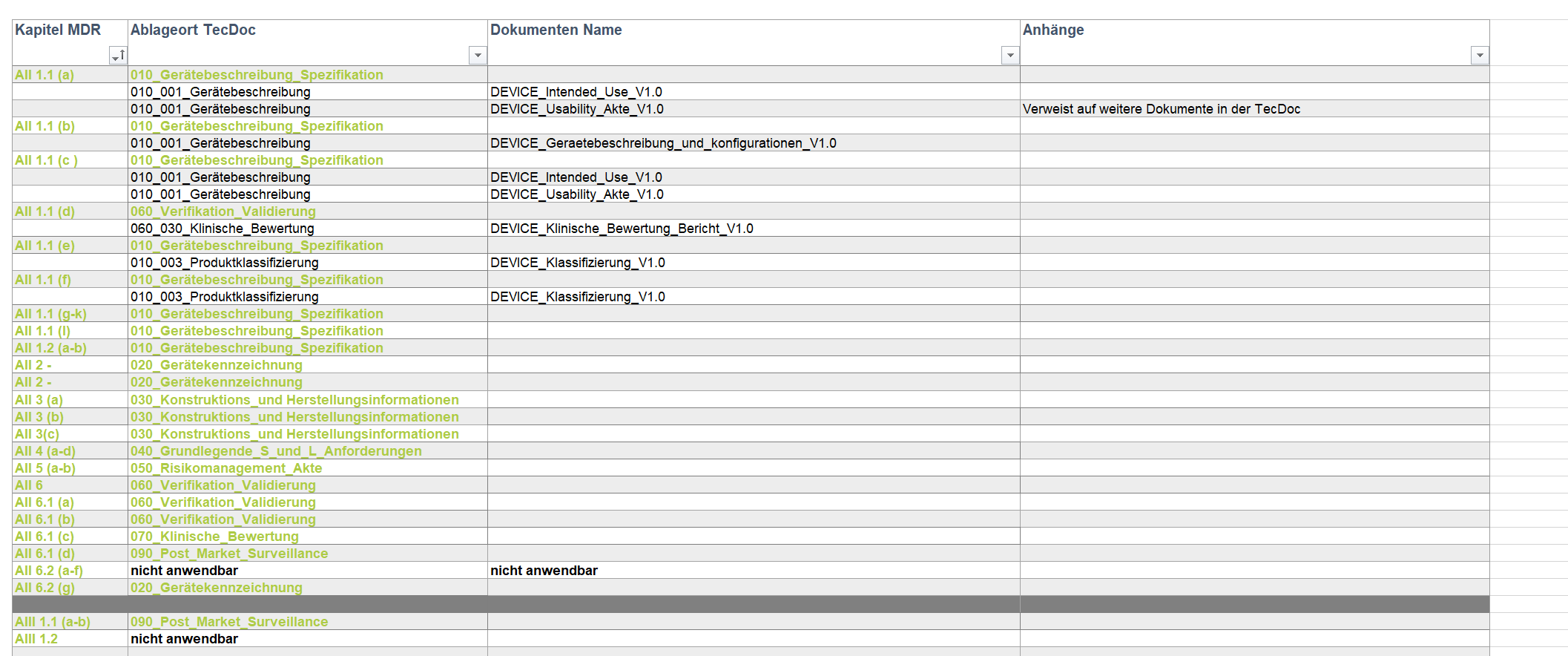
Implementierung und Anwendung eines Software-Entwicklungsprozess nach IEC 62304 – Projekt im Tandem
IPP Notizen aus der Praxis
Ausgangslage
Der Kunde ist Hersteller von individuell konfigurierbaren Behandlungseinheiten. Da diese Software beinhalten, musste die Software-Lebenszyklus-Norm IEC 62304 Anwendung finden. Die Norm unterscheidet die Software Klasse A, B und C, wobei der Dokumentationsaufwand für Klasse B und C etwas höher ist als für Klasse A. Die Behandlungseinheiten setzen sich aus mehreren Hauptkomponenten zusammen, die jeweils eigene Software beziehen. Somit konnte jede Software für sich klassifiziert werden. Die Entwicklung und Dokumentation muss einem Prozess folgen, der für die Software gemäß IEC 62304 implementiert und nach Prozess-Einführung angewendet wurde.
Projektziel
-
- Einführung und Implementierung des Software-Entwicklungsprozess nach IEC 62304
- Anwendung des Prozesses für alle Software-Komponenten
- Erstellung eines vollständigen Nachweises zur IEC 62304
Durchführung
Prozesserweiterung
Mit Hilfe einer GAP-Analyse (“Lücken-Analyse”) durch IPP konnte der Kunde den bestehenden Entwicklungsprozess um die softwarespezifischen Prozessschritte ergänzen. Somit konnte dieser sein Flow-Chart für die Prozess-Ansicht optimieren und erweitern.
Vorlagen und Dokumente
Anhand des Prozesses hat IPP entsprechende Vorlagen erstellt und durch den Kunden freigegeben.
Dies war durch die „IPP-Bibliothek“ aus Easy13485 möglich, wodurch im Projekt schnell Fahrt aufgenommen werden konnte. Anschließend konnten die angepassten Vorlagen mit entsprechenden Inhalten gefüllt werden, sodass in sich logische Dokumente gemäß Prozessreihenfolge entstanden sind. Durch den Team-Spirit getrieben, entstand eine verlässliche Zusammenarbeit zwischen dem Kunden und IPP sowie mit einem externen Software-Entwickler, der umfangreiche Dokumente mit softwarespezifischem Inhalt pünktlich lieferte.
Nachweis der IEC 62304
Durch die neu erstellten Dokumente, konnte der Prozess erfolgreich eingeführt und die IEC 62304 für Software Klasse A und Klasse B vollständig erfüllt und nachgewiesen werden.
Ergebnis
Durch die gemeinsame Arbeit konnte der Kunde das Projekt “Prozess-Einführung für Software gemäß IEC 62304 und Anwendung” in relativ kurzer und fristgerechter Zeit beenden.
Dabei war der rote Faden des Prozesses immer erkennbar und für alle Beteiligten ersichtlich.
Letztendlich hat das anstrengende Projekt allen Spaß gemacht.
Lesson-Learned
Ausblick
Die durchdachte und geplante Vorgehensweise wird vermutlich auch eine Signalwirkung auf die Arbeit in zukünftigen Projekten des Kunden haben. Über dieses Projekt hinaus möchte der Kunde weiterhin für neue Projekte durch IPP unterstützt werden, da dieser festgestellt hat, dass komplexe Projekte im Tandem besser funktionieren als allein. Das positive Feedback des Kunden freut uns als IPP sehr und wir sind gespannt auf die weiteren Projekte!
Weitere interessante Artikel
Umstellung Klasse IIa TecDoc auf MDR – erfolgreiche Einreichung einer “Pilot-TecDoc”
Technische Dokumentation nach MDR für extraorale Röntgengeräte
Notfallmaßnahmen für MDR Audit Abweichungen: Wie wir unserem Kunden geholfen haben