Technische Dokumentation nach MDR für die Erprobung eines neuen XRay-Sensors für extraorale Röntgengeräte
IPP Notizen aus der Praxis:
Ziel: CE-Konformität nach MDR / Medizinprodukte Verordnung 2017/745 für ein extraorales Röntgengerät
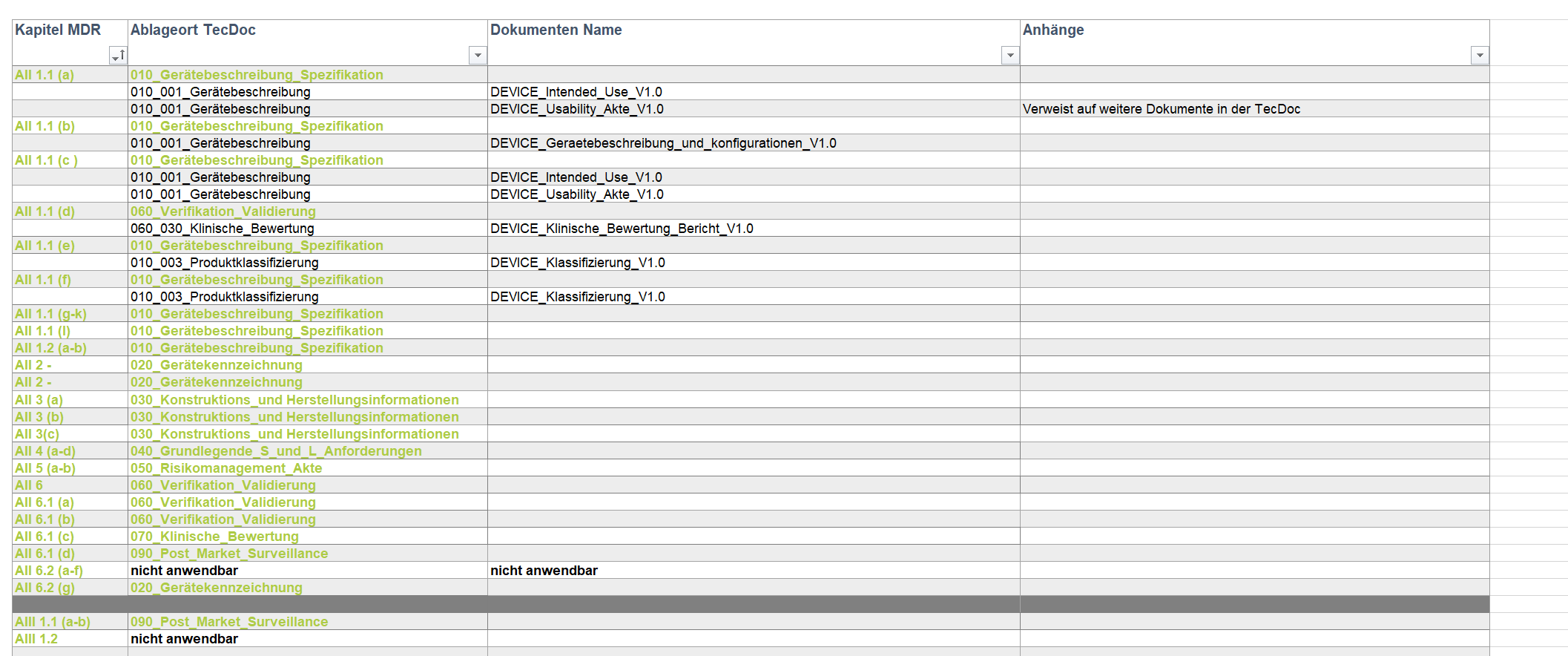
Aufgabe: Als externer Experte hat IPP in diesem Projekt die Planung sowie Umsetzung im Fachbereich Design Control übernommen. Dies umfasste die komplette Erstellung der Technischen Dokumentation (TecDoc) bzw. Produktakte nach MDR (Medical Device Regulation, 2017/745), um für die Erprobung eines neuen Sensors in einem extraoralen Röntgengerät eine CE-Konformität zu erhalten.
Zeitlicher Rahmen: Für dieses Projekt standen insgesamt 9 Monate zur Verfügung.
Durchführung
- Erstellung der TecDoc nach MDR in Abstimmung mit der Projektleitung
- Berücksichtigung der Prozesse des Kunden bei der Erstellung der Dokumentation
- Vorbereitung und Durchführung der Review Termine
- Kontinuierlicher Austausch mit der Approbation bzw. dem gesamten Projektteam des Herstellers
- Erstellung eines Medical Device Files, worüber der Kunde die Vollständigkeit der TecDoc eigenständig prüfen und Benannten Stellen vorweisen kann
Ergebnis
Die Technische Dokumentation nach MDR konnte mit unserer Unterstützung und der durchweg positiven Zusammenarbeit zwischen allen Beteiligten, intern sowie extern, termingerecht sowie prozess- und normkonform erstellt werden. Der Kunde konnte somit die Erprobung des neuen Sensors erfolgreich starten.
Fragen?
Sie benötigen Unterstützung oder Beratung zum Thema Technische Dokumentation von Medizinprodukten durch unsere Experten? Sie erreichen uns telefonisch unter : +49 911 36069710 sowie per Email unter kontakt(at)ipp-nbg.com
[contact-form-7 id=”148714″ title=”Kontaktformular 1″]
Bei der MDR handelt es sich um die Medical Device Regulation / Medizinprodukt Verordnung 2017/745 der EU, an die sich die Hersteller von Medizinprodukten halten müssen.