In welcher Sprache muss eine Technische Dokumentation nach MDR vorliegen?
Die Sprache der Technischen Dokumentation
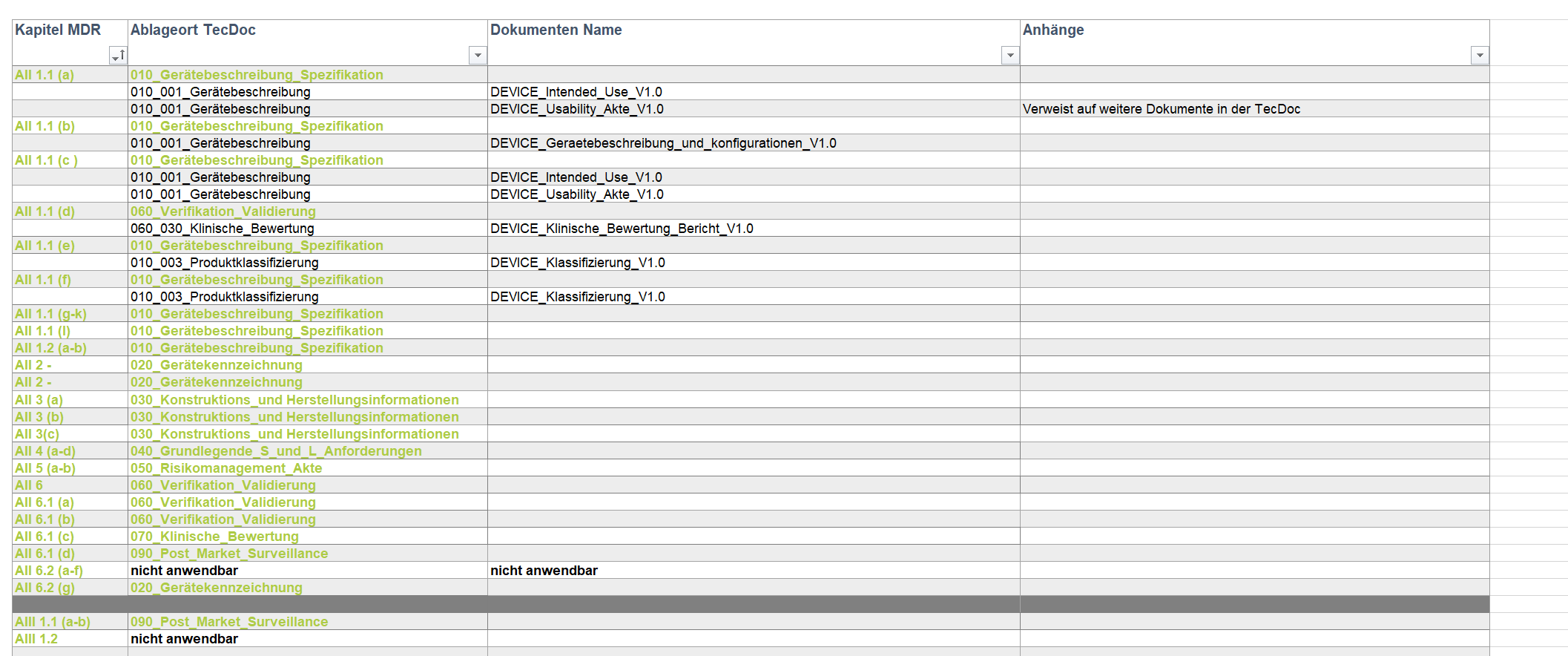
Die technische Dokumentation, Technical File, Produktakte oder auch einfach nur TecDoc ist das zentrale Nachweisdokument beim Konformitätsbewertungsverfahren zur CE-Kennzeichnung für Medizinprodukte. Dementsprechend muss auch die Sprache von diesem wichtigen Dokument entsprechend richtig ausgewählt sein.
Ausgangslage:
Dies ist vor allem für Hersteller sowie Lieferanten von Medizinprodukten mit der Konstellationen „OEM“ „PLM“ interessant.
Als Hersteller entwickeln und produzieren sie selbst. Sie stellen selbst die Konformität aus, wenn die Erfüllung der Kriterien zur Konformität schriftlich nachgewiesen ist.
Sie sind auch Hersteller, wenn Sie ein Medizinprodukt oder Komponenten dafür von einem OEM kaufen und unverändert mit Ihrem eigenen Typenschild in Verkehr bringen. Oder Sie bringen ein fertig entwickeltes Medizinprodukt von einem OEM nach eigenen Wünschen modifiziert sowie mit einem eigenen Typenschild in Verkehr.
Zusammenfassend muss in allen genannten Fällen eine technische Dokumentation vorliegen.
Was ist aber, wenn Ihr OEM z.B. in China sitzt und “Ihr(e)” Medizinprodukt(e) oder Komponente nur eine chinesische TecDoc besitzt? Müssen sämtliche Dokumente übersetzt werden?
Es kommt darauf an in welchem Mitgliedsstaat Ihre Benannte Stelle (BS) niedergelassen ist und was sie entsprechend verlangt.
Die BS kann verlangen, dass alle oder bestimmte Unterlagen, darunter die technische Dokumentation, Audit-, Bewertungs- und Kontrollberichte, im Zusammenhang mit den […] genannten Verfahren (Anmerkung ipp: Konformitätsverfahren) in einer oder mehreren von diesem Mitgliedstaat festgelegten Amtssprachen der Union bereitgestellt werden muss. Wird dies nicht verlangt, so müssen diese Unterlagen in einer Amtssprache der Union vorliegen, mit der die Benannte Stelle einverstanden ist. (siehe MDR 2017/745 Artikel 52 Konformitätsbewertungsverfahren Absatz (12))
Ansonsten müssen Hersteller der zuständigen Behörde auf deren Ersuchen hin alle Informationen und Unterlagen, die für den Nachweis der Konformität des Produkts erforderlich sind, in einer von dem betreffenden Mitgliedstaat festgelegten Amtssprache der Union aushändigen. (siehe MDR 2017/745 Artikel 10 Allgemeine Pflichten der Hersteller Absatz (14); siehe auch Artikel 11 Bevollmächtigter Absatz d)
Also klären Sie vorher mit Ihrer BS ab, in welcher Sprache die Unterlagen vorliegen sollen.
Benötigen Sie weitere Hilfe bei der Erstellung, Anpassung oder Umstellung der Technischen Dokumentation? Reichen Ihre eigenen Ressourcen nicht mehr aus?
Unsere fachkompetenten Experten helfen Ihnen schnell sowie effizient in allen Bereichen der Technischen Dokumentation.
» Sprechen Sie uns an, wir erarbeiten Ihnen Ihr individuelles Angebot!