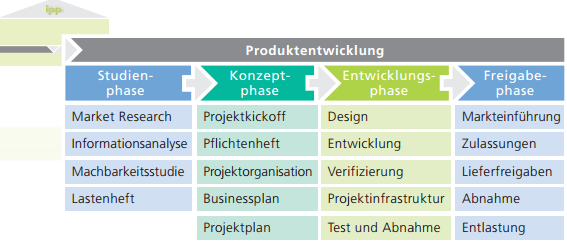
Medizinprodukt: Verifizierung und Validierung nach DIN EN 13060 und ISO 15883 mit Technischer Dokumentation
Die Verifizierung und Validierung
IPP Notizen aus der Praxis:
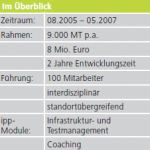
Innerhalb von zwei Jahren sollte ein Prototyp eines Reinigungs-Desinfektionsgeräts für Dentalinstrumente termingerecht auf den Markt gebracht werden. Dementsprechend muss auch eine Verifizierung und Validierung gemäß der Normen erfolgen.
Hierzu mussten Prototyp, Nullserien- und Seriengerät inklusive Dokumentation gegen Systemtestplan und Normen verifiziert und validiert werden. Die Entwicklung und Produktion erfolgte international an unterschiedlichen Standorten mit über 30 Projektmitarbeitern.
Ziel war es, die verantwortlichen internen Entwickler hinsichtlich der Verifizierungs- sowie Validierungsaktivitäten und Technischer Dokumentation zu entlasten, damit der Fokus weiterhin auf die Entwicklungsarbeit liegen konnte.
Durchführung Verifizierung und Validierung
- Verantwortlichkeit für Planung, Koordination sowie Dokumentation der Aktivitäten der Verifizierung und Validierung im Projekt (Testplanung / Testdurchführung / Testreporting) nach DIN EN 13060, ISO 15883 und DIN EN 60529
- Reporting der Versuchsergebnisse an den Projektleiter
- Vorschlag und Planung von Arbeitspaketen zur Verbesserung, die sich aus der Verifikation ableiten, sowie Umsetzung dieser Arbeitspakete
- Unterstützung im Bereich der Risikoanalyse
- Normgerechte Dokumentation aller Ergebnisse nach MDD 93/42/EWG
Ergebnis
Das Erreichen des Gesamtprojektziel (Termin, Budget, Qualität).
Alle Aktivitäten der Verifizierung und Validierung wurden inklusive Dokumentation termingerecht abgeschlossen.
Das gesamte Innovations- Knowhow verbleibt beim Hersteller. Alle Akten können intern weitergepflegt werden.
Fragen?
Sie benötigen Unterstützung oder Beratung zum Thema Technische Dokumentation von Medizinprodukten durch unsere Experten? Sie erreichen uns telefonisch unter : +49 911 36069710 sowie per Email unter kontakt(at)ipp-nbg.com