Moritz Nickel

Verifizierung von Medizinprodukten
Manager
Medical Device Expert & Project Management
Moritz Nickel ist ein erfahrener Spezialist im Bereich Verifizierung von Medizinprodukten und technischer Dokumentation mit umfassender Expertise in der Medizintechnik. Er hat seinen Master of Science in Medizintechnik (Medical Engineering) an der Friedrich-Alexander-Universität Erlangen-Nürnberg abgeschlossen. Sein Schwerpunkt lag dabei auf medizinischer Produktionstechnik, Gerätetechnik und Prothetik.
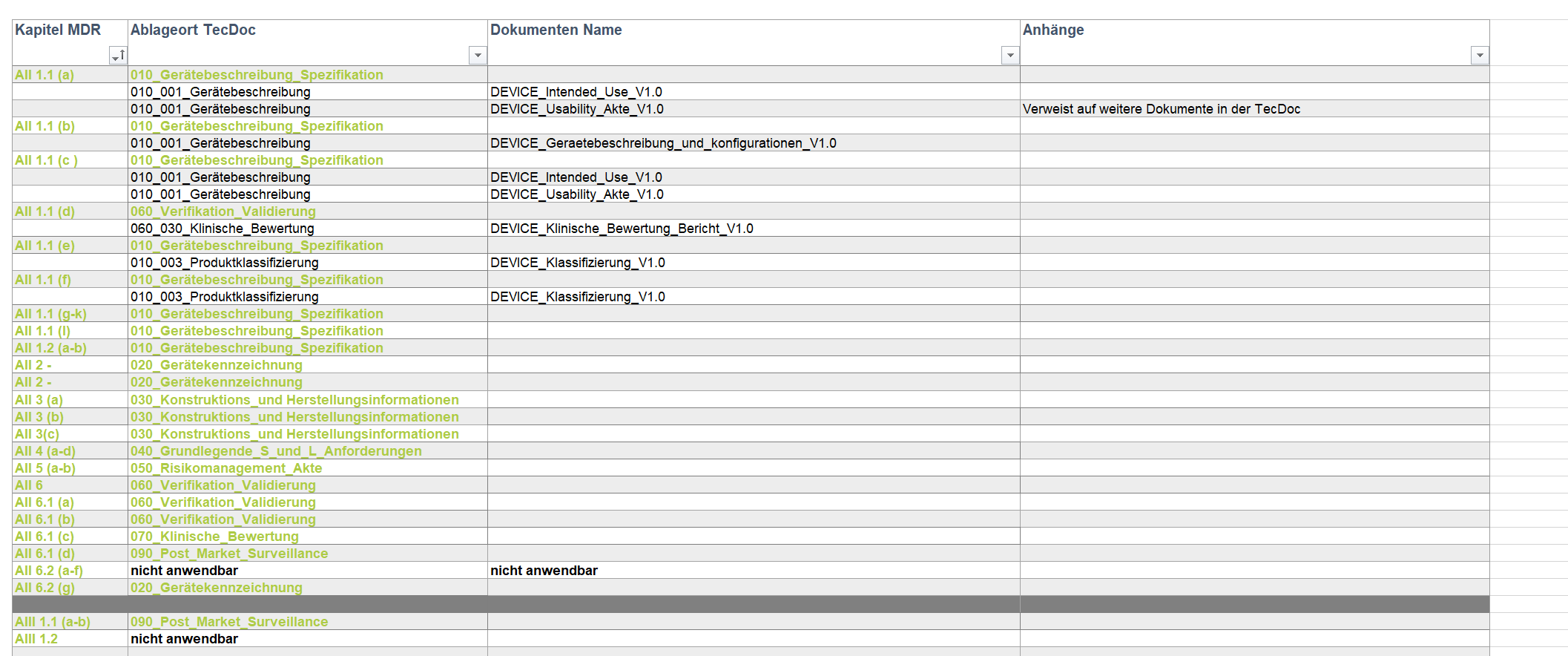
Durch seine langjährige berufliche Erfahrung hat sich Moritz Nickel als gefragter Experte für die Erstellung technischer Dokumentationen nach MDR etabliert. Sein Fokus liegt dabei sowohl auf der Umstellung bestehender Produktakten als auch auf der projektbegleitenden Neuerstellung nach MDR.
Mit der erfolgreichen Absolvierung des Lehrgangs “Expert Medical Software (TÜV)” hat sich Moritz Nickel zusätzlich auf Usability Engineering nach IEC 62366-1, Risikomanagement nach ISO 14971 und die CE-Kennzeichnung von Medical Device Software (MDSW) spezialisiert. Er verbindet agile Entwicklungsmethoden mit den geltenden regulatorischen Vorgaben. Dabei unterstützt er Unternehmen bei der sicheren und konformen Entwicklung medizintechnischer Produkte.
Während seiner Karriere war Moritz Nickel unter anderem als Design Control Projektleiter bei Sirona Dental Systems tätig, wo er die Erstellung und Koordination der zulassungsrelevanten Dokumentation für unterschiedliche Produkte verantwortete. Vor dieser Rolle war er in der Erweiterung eines Softwaretools zur Systemteststeuerung involviert und sammelte umfangreiche Erfahrungen in der Digitalisierung und Migration von Produktakten in ein ALM-System. Hierbei eignete er sich fundiertes Fachwissen in Softwareentwicklung, Prozess- und Projektdokumentation sowie im Umgang mit dem ALM- System Polarion an.
Aktuell ist Moritz Nickel als Teilprojektleiter an der Entwicklung eines extraoralen Röntgengeräts bei Sirona Dental Systems beteiligt. Dabei verantwortet er die Erstellung einer MDR-konformen technischen Dokumentation und bereitet diese auch für die 510(k)-Zulassung vor. Zudem unterstützt er die Entwicklung bei sämtlichen Design Control Aufgaben und sorgt für die Einhaltung regulatorischer Vorgaben.
Seine Fachkompetenz in den Bereichen Requirements Engineering (IEC 62304), technische Dokumentation und Regulatory Projektmanagement macht ihn zu einem unverzichtbaren Partner für die erfolgreiche Verifizierung und Zulassung von Medizinprodukten.
Moritz Nickel ist Ihr Spezialist für die Verifizierung von Medizinprodukten, technische Dokumentation nach MDR, Requirements Engineering (IEC 62304) sowie Regulatory Projektmanagement am Standort Nürnberg in Bayern.