Technische Dokumentation nach MDR für die Erprobung eines neuen XRay-Sensors für extraorale Röntgengeräte
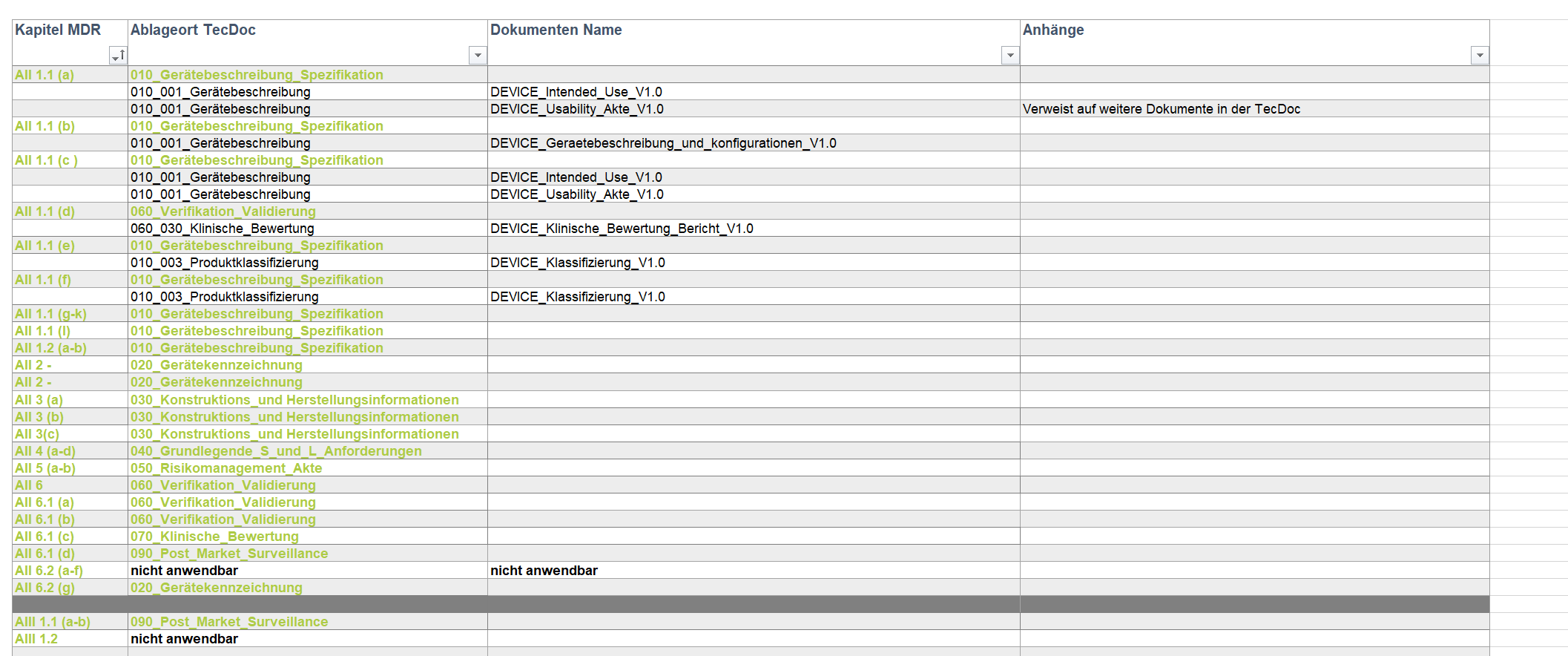
ipp. unterstützte die Erstellung der Technischen Dokumentation nach MDR für einen neuen XRay-Sensor. Innerhalb von neun Monaten wurde eine normkonforme Produktakte erstellt, die die CE-Konformität nach MDR 2017/745 für die Erprobung des Sensors ermöglichte.